

{ DOWNLOAD AS PDF }
ABOUT AUTHORS
1Veerendra Kr. Gautam*, 2Mohamad. Irfan
1Executive in Drug Regulatory Affairs Department; East African (India) Overseas
1 120 Suncity Business Tower,Sector-54, Gurgaon-122002 (Haryana).
2Research Associate; Jubilant Chemsys Ltd.
2D-12, Sector 59, Noida , Uttar Pradesh, India
1dra.veerendra.gautam@gmail.com;
1viren.gautam.dra@gmail.com 2mohd.irfan.ivar@gmail.com
Abstract : Dossier is a collection of documents on the particular subjects. Any preparation of pharmaceutical product for human use go through the process of reviewing and assessing the dossier of pharmaceutical product which contains details information about administrative, quality, non-clinical and clinical data. This process is governed and permitted by Drug Regulatory Authority of a particular country and process is called as NDA in USA, MAA in EU and other countries as simply Registration Dossier. There are basically two formats for dossier preparation i.e. ICH-CTD and ACTD. ICH-CTD followed by ICH countries as well as low economical or developing countries where as ACTD is followed by ASEAN countries. ACTD act as bridge between regulatory requirements of developed and developing countries. Also if both guidelines of CTD and ACTD can be harmonized then differences and variation between both guidelines can be minimized.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2527
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 5, Issue 10 Received On: 07/06/2017; Accepted On: 08/06/2017; Published On: 01/10/2017 How to cite this article: Gautam VK, Mohamad I; A Study of procedures for Dossier Preparation and their marketing authorisation in different countries of selected drug(s); PharmaTutor; 2017; 5(10); 8-22 |
INTRODUCTION
Dossier [1-4] The word 'Dossier' has the english meaning as a collection or file of documents on the particular subject, especially a file containing detailed information about a person or a topic. Any formulation is prepared for human use i.e. designated to modify or explore physiological systems or pathological states for the benefit of the recipient is called as “Pharmaceutical product for human use”. Process of critiquing and assessing the dossier of pharmaceutical product containing its detailed about administrative, chemistry, preclinical & clinical information and the permission granted by the regulatory agencies of a country with a view to support its marketing or approval in a country is called as “Marketing approval or Registration” ,“Marketing Authorization or “ Product Licensing”.
“Registration Dossier” of the pharmaceutical product is a document that contains all technical data (administrative, quality, nonclinical, and clinical) of a pharmaceutical product to be approved / registered / marketed in a country. It is more commonly called as New Drug Application (NDA) in the USA or Marketing Authorization Application (MAA) in European Union (EU) and other countries as simply Registration Dossier.
The International Conference on Harmonization (ICH) process has considerably harmonized on the organization of the registration of documents with the issuance of the Common Technical Document (CTD) guideline. This recommended format in the CTD guideline for registration applications has become widely accepted by regulatory authorities both within and beyond the ICH Regions.
Thus dossier is a file document that has to be submitted based on the requirement of the drug approval/ market authorization process. It is a comprehensive scientific document used to obtain worldwide licensing approval/ market authorization of a drug by diverse health authorities. Its creations, processing, compilation & dispatch to the field by a regulatory affairs department, is dependent upon many interrelated activities, the filling and authorization process in the emerging markets will be depends upon the region.
Globalization of the pharmaceutical industry has created the need to harmonize the recommendations for the development of new pharmaceuticals, as well as the regulatory requirements of various countries. Thus, a common format of submission will help in overcoming these hurdles. Through ICH process, the CTD’s guidance have been developed for Japan, European Union, and United States. Almost Most of the countries have adopted the CTD format.
COMMON TECHNICAL DOCUMENT (CTD) [5]
CTD is a set of specification for application dossier for the registration of Medicines and designed to be used across Europe, Japan and the United States. It was developed by the European Medicines Agency (EMA, Europe), the Food and Drug Administration (FDA, U.S.) and the Ministry of Health, Labor and Welfare (Japan). The CTD is maintained by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. The agreement to assemble all the quality, safety and efficacy information in a common format has revolutionized the regulatory review processes.
General Consideration
- CTD is -
* Only a harmonized format for submission of information to relevant regulatory
authorities.
* Template for presenting data in the dossier.
* A guideline that merely indicates an appropriate format for the data that have
been acquired.
- CTD is not -
* A statement of data for application of data.
* A guideline that intends to indicate what studies are required.
* Define the content.
- CTD should be -
*Have clear and unequivocal information.
*Have style & font size that is large enough to be easily readable.
*Follow the ICH guidelines for:
Document pagination and segregation.
Submission requirements/ methodology for CTD.
* Contained all abbreviation that are used & be listed at the end of the dossier.
* Give proper information of source of bulk drug(s) for manufacturing finished
formulation.
Regulation & regulatory bodies of CTD [5]
1) The regulation under Drugs and Cosmetics Act & Rules 122A, 122B and 122D and further Appendix I, IA and VI of Schedule Y, describe the information required for approval of an application to import or manufacture of new drug for marketing.
2) Every country has its own regulatory authority, which is responsible to enforce the rules and regulations and issue guidelines for drug development, licensing, registration, manufacturing, marketing and labeling of pharmaceutical products.
3) Almost all the independent countries of the world have their own regulatory authorities.
Evolution of CTD [6]
Effort over the past 15- 20 years by ICH of technical requirements for "registration of pharmaceutical for human use" have resulted in a uni-field dossier for drug applications. CTD was officially signed off in November 2000, at 10th anniversary of ICH; San Diego, California.
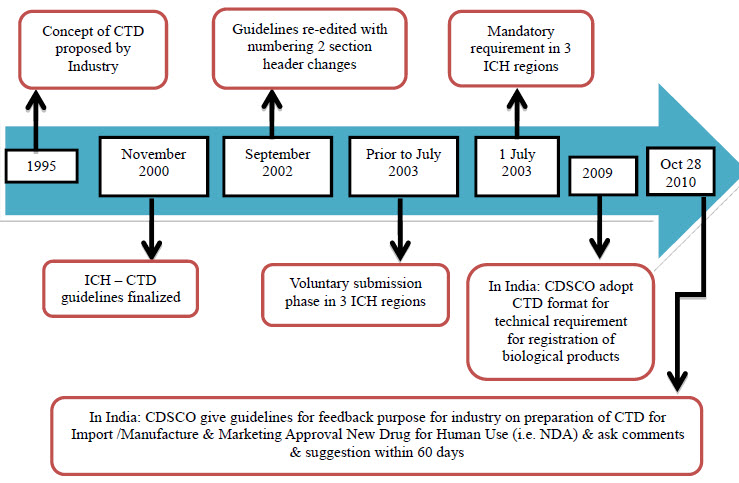
Fig 1 : Evolution of CTD
The following are the examples of a few of them (Regulatory Authority):
Table 1 : List of countries and their Regulatory Authority (ICH-CTD)[4]
|
Country |
Regulatory Authority |
|
Australia |
Therapeutic Goods Administration (TGA) |
|
Argentina |
National administration of Drugs, Food & Medical Technology (ANMAT) |
|
Armenia |
Drug & medical Technology Centre, Ministry of Health |
|
Austria |
Federal ministry for Health |
|
Brazil |
National Agency for Sanitary Vigilancia (ANVISA) |
|
Belgium |
Federal public services( FPS) Health |
|
Bolivia |
Ministry of Health and Sports |
|
Bulgaria |
Bulgarian Drug Agency (BDA) |
|
Canada |
Health Canada |
|
China |
State Food and Drug Administration (SFDA) |
|
Colombia |
National Institute of Food and Drug monitoring( INVIMA) |
|
Croatia |
Ministry of Health and Social Welfare |
|
Denmark |
Danish Medicines Agency |
|
Ecuador |
Ministry of Public Health |
|
Egypt |
Ministry of Health & population |
|
Estonia |
State Agency of Medicines |
|
Europe |
European Medicines Agency (EMEA) |
|
Fiji |
Ministry of Health |
|
Finland |
Finnish Medicines Agency |
|
France |
French Agency for Sanitary Safety of Health Products, ministry of Health |
|
Germany |
Federal Institute for Drugs and Medical Devices |
|
Georgia |
Ministry of Labor, Health and Social Affairs of Georgia |
|
Greece |
National Organization for Medicines (EOF) |
|
Guam |
Department of Public Health and Social Services |
|
Hong Kong |
Department of Health: Pharmaceutical Services |
|
Hungary |
National Institute for Pharmacy |
|
Iceland |
Icelandic Medicines Agency |
|
India |
Central Drug Standard Control Organization (CDSCO) |
|
Ireland |
Irish Medicines Board |
|
Italy |
Italian Pharmaceutical Agency |
|
Jamaica |
Ministry of Health |
|
Japan |
Ministry of Health, Labour & Welfare(MHLW) |
|
Jordan |
Ministry of Health |
|
Kenya |
Ministry of Health |
|
Latvia |
State agency of medicines |
|
Lithuania |
State Medicines Control Agency |
|
Maldives |
Ministry of Health and Family |
|
Malaysia |
National Pharmaceutical Control Bureau, Ministry of Health |
|
Mauritius |
Ministry of Health and Quality of Life |
|
Namibia |
Ministry of Health and Social Services |
|
Nepal |
Ministry of Health and Population |
|
New Zealand |
Medicines and Medical Devices Safety Authority (MEDSAFE) |
|
Nigeria |
National Agency for Food and Drug Administration and Control (NAFDAC) |
|
Norway |
Norwegian Medicines agency |
|
Panama |
Ministry of Health |
|
Paraguay |
Department of Health: Pharmaceutical Services |
|
Poland |
Ministry of Health & Social Welfare |
|
Portugal |
The National Institute of Pharmacy and Medicines |
|
Romania |
National Medicines Agency (ANM) |
|
Russia |
Ministry of Health and Social Development |
|
Senegal |
Ministry of Health & Prevention |
|
Serbia |
Medicines and Medical devices Agency (ALIMS) |
|
Singapore |
Center for Pharmaceutical Administration Health Sciences Authority |
|
South Korea |
Food and Drug Administration |
|
South Africa |
Medicines Control Council (MCC) |
|
Spain |
Medicines and Health Product Agency (AEMPS) |
|
Switzerland |
Swiss Agency for Therapeutic Products (SWISSMEDIC) |
|
Taiwan |
Department of Health |
|
Tanzania |
Ministry of Health and Social Welfare |
|
Thailand |
Ministry of Public Health |
|
UK |
Medicines and Healthcare Products Regulatory Agency (MHRA) |
|
USA |
Food and Drug Administration (FDA) |
|
Uruguay |
Ministry of Public Health |
|
Venezuela |
Ministry of Public Health |
|
Vietnam |
Ministry of Health |
|
Yemen |
Ministry of Public Health and Population |
|
Zimbabwe |
Medicine Control Authority of Zimbabwe (MCAZ) |
INTERNATIONAL ORGANIZATIONS [7-10]
Some of the international regulatory agencies and organizations which also play essential role in all aspects of pharmaceutical regulation related to drug product registration, manufacturing, distribution, price control, marketing, research and development and intellectual property protection.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
World Health Organization (WHO) [7]
When diplomats met to form the United Nations in 1945, one of the things they discussed was setting up a global health organization.
Constitution of the World Health Organization : The Constitution was adopted by the International Health Conference held in New York from 19 June to 22 July 1946, signed on 22 July 1946 by the representatives of 61 States and entered into force on 7 April 1948 - a date we now celebrate every year as World Health Day.
Pan American Health Organization (PAHO)[8]
The Pan American Health Organization (PAHO), founded in 1902, is the world’s oldest international public health agency. It provides technical cooperation and mobilizes partnerships to improve health and quality of life in the countries of the Americas. PAHO is the specialized health agency of the Inter-American System and serves as the Regional Office for the Americas of the World Health Organization (WHO). Together with WHO, PAHO is a member of the United Nations system.
World Trade Organization (WTO) [9]
The WTO is a rules-based, member-driven organization - all decisions are made by the member governments, and the rules are the outcome of negotiations among members.
International Conference on Harmonization (ICH) [10]
ICH’s mission is to make recommendations towards achieving greater harmonization in the interpretation and application of technical guidelines and requirements for pharmaceutical product registration, thereby reducing or obviating duplication of testing carried out during the research and development of new human medicines.
Launched in 1990, ICH is a unique undertaking that brings together the drug regulatory authorities and the pharmaceutical industry of Europe, Japan and the United States.
Regulatory harmonization offers many direct benefits to both regulatory authorities and the pharmaceutical industry with beneficial impact for the protection of public health.
PROGRESS IN INTERNATIONAL HARMONIZATION
There have been enormous changes in the technical and scientific requirements for the dossier, as the work of the ICH has continued to devise and approve new guidelines in the categories of Quality, Safety, Efficacy, and Multidisciplinary. The CTD harmonized structure and modular format for new medicinal product registration files that were adopted in San Diego, is now the obligatory format in the European Union (EU), Japan, Canada, Switzerland, and Australia. It is the recommended format in the United States.
THE CTD—A COMMON FORMAT, NOT A HARMONIZED CONTENT FOR SUBMISSIONS
Enormous efforts have been expended by the staffs of the regulatory agencies and the pharmaceutical industry in the work of the ICH, and this has achieved a remarkable degree of harmonization in many scientific and technical areas of the dossier. Despite this there are still national differences in the content of submissions not only in Module 1, the administrative and prescribing information, but also in other areas of the dossier. These arise from differences in regulatory practice and procedures, different practices of medicine and pharmacy, and differences in access to diagnostic and therapeutic procedures. We are, however, still far from a genuinely global single registration dossier.
INCREASED COOPERATION BETWEEN AGENCIES BASED ON ICH
Mutual Recognition Agreements between agencies in relation to Good Manufacturing Practice (GMP) are in operation between the EU and Canada, Australia, New Zealand, Switzerland, and Japan. Arrangements exist between many countries (including the ICH members) for exchange of pharmacovigilance and defect information.
The confidentiality arrangements between the EU and the FDA now allow for exchange of information on legal and regulatory issues, scientific advice, orphan drug designation, inspection reports, marketing authorization procedures, and post-marketing surveillance. In September 2004, the European Medicines Agency (EMEA) and the Food and Drugs Administration (FDA) instituted a pilot program of parallel scientific advice meetings for sponsors to obtain advice on scientific issues during the development phase of new medicinal products. Orphan indication products and pediatric products have been targeted in particular. Bilateral and trilateral collaboration has increased in 2008. Health Canada has agreed to exchange information with the European Commission and EMEA about the authorization and safety of drugs. Canada and Australia have started their parallel review project for biologicals (originally launched in 2006). All of this has only been possible based on the prior work that has been done in harmonization of regulatory requirements and in the development of the CTD format of the dossier in ICH.
SPREADING THE ICH MESSAGE—THE ICH GLOBAL COOPERATION GROUP
The ICH-affiliated and other developed countries, which have adopted the CTD format (the United States, EU, Japan, Canada, Switzerland, and Australia), comprise in total approximately 15% of the current (2008) world population of 6650 million people. The ICH Global Cooperation Group (GCG) was formed on March 11, 1999 as a subcommittee of the ICH Steering Committee. Its purpose is to make information available on ICH, ICH activities, and any ICH guideline to a wider group of countries.
Regional Harmonization Initiatives
A number of regional harmonization initiatives (RHIs) have been set up where a geographic grouping of countries harmonizes technical and scientific requirements and in some cases the format of submissions for member countries. These groups have been invited to designate permanent representatives to the GCG. They currently comprise:
Asia-Pacific Economic Cooperation (APEC): 21 countries in the Asia-Pacific region.
Association of Southeast Asian Nations (ASEAN): Brunei Darussalam, Cambodia, Indonesia, Laos, Malaysia, Myanmar, Philippines, Singapore, Thailand and Vietnam.
Gulf Cooperation Countries (GCC): Saudi Arabia, Kuwait, United Arab Emirates, Oman, Bahrain, Qatar and Yemen.
Pan American Network on Drug Regulatory Harmonisation (PANDRH): Argentina, Barbados, Bolivia, Brazil, Chile, Colombia, Costa Rica, Cuba, Guatemala, Jamaica, Mexico, Panama, Trinidad and Tobago and Venezuela.
South African Development Community (SADC): Angola, Botswana, Democratic Republic of Congo, Lesotho, Malawi, Mauritius, Madagascar, Namibia, Seychelles, South Africa, Swaziland, Tanzania, Zambia and Zimbabwe.
The regional groups review the applicability of the ICH guidelines in their own specific countries. Particular topics of interest include the ICH stability guideline, GMP guidances, requirements for bioavailability and bioequivalence studies, clinical trials, export/import of medicines, traditional medicines, and market surveillance.
The following are the various regions as per regulatory guidelines having CTD structure / format for registration product-[11-13]
* ASEAN Region (ACTD Format): Brunei, Cambodia, Indonesia, Lao People’s Democratic Republic, Malaysia, Myanmar, Philippines, Singapore, Thailand and Vietnam.
* African Region (CTD Format): Nigeria, Kenya, South Africa, Zimbabwe, Tanzania, Ethiopia, Namibia, & Mauritius.
* CIS Region (CTD Format) (country specific resemble to CTD): Maldova, Russia, Ukraine, Georgia, Kazakhstan, Uzbekistan.
* WHO & India (CTD Format)
* LATAM region (Country specific): Mexico, Panama, Venezuela, Chile, Costa-Rica, Brazil, Dominican Republic & Jamaica.
* GCC Region (CTD Format) and Australia (ICH-CTD Format)
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
The registration procedure is differs from region to region. Thus some follow the ICH guidelines and some WHO guidelines for the registration of the drug product. But some regions have the country specific guidelines for registration of the FPP. Drug regulatory affairs in pharmaceutical industries have mandated two types of dossier namely CTD (Common Technical Dossier) and ACTD (ASEAN Common Technical Dossier). Regulated pharmaceutical markets (eg. USA, Europe) require submission of dossier in CTD format which require clinical trial and bioequivalence studies. As against this, semi-regulated pharmaceutical markets (South East Asian) require ACTD format which does not require exhaustive details like CTD. All of these guidelines will consider the safety, quality and efficacy of Finished Pharmaceutical Product (FPP).
ORGANIZATION OF ICH-CTD FORMAT [14-18]
The CTD is organized into five modules. Module 1 is region-specific. Modules 2, 3, 4 and 5 are be common for all regions. Conformance with ICH guidelines should ensure that these four modules are provided in a format acceptable to WHO and regulatory authorities. An overview of module contents for a multisource product in greater details.
The five Modules are:
- Module 1: Administrative and prescribing information
- Module 2: Overview and summary of module 3 to 5
- Module 3: Quality (Pharmaceutical documentation)
- Module 4: Safety (Toxicology/ Non-clinical studies)
- Module 5: Efficacy (Clinical studies)
The diagram below represents the above information as a modular structure which is known as CTD Triangle.
The modular structure of ICH-CTD shows that Module 1 is not a part of CTD, it contain only the regional information or administrative information of the one who right to file the dossier for getting market authorization.
The modular structure is detailed with the help of technical data of common technical document (CTD) which is mentioned in all module as module of contents.
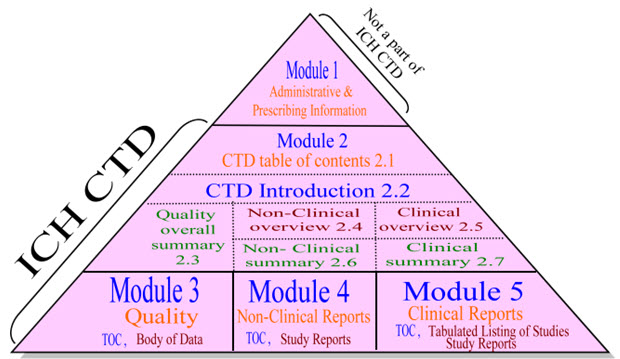
Fig 2 : Modular Structure of ICH CTD
This format is explained with an examples of two selected countries Zimbabwe and Australia from different continents which follow the ICH-CTD format for dossier preparation for selected drug(s) Regorafenib and Roflumilast respectively.
CATEGORIES OF PHARMACEUTICAL PRODUCT THAT CAN BE SUBMITTED AS A CTD REGISTRATION FILE
Most categories of product can be submitted in all of the ICH regions as a CTD registration file. This includes the following:
- New drug substance (NCE) products
- New biological/biopharmaceutical products
- Radiopharmaceuticals
- Phytopharmaceuticals (herbal medicines)
In the case of the United States, CTD registration files may also be submitted for generic and OTC products and this format would be obligatory in the European Union for these products.
DIFFERENCES IN CTD CONTENT BETWEEN THE ICH REGIONS
As mentioned in the Preface, the CTD is a harmonized format for registration files; however, the content is not yet completely harmonized. There are still national or regional differences in the content of submissions—not only in Module 1 but also in other parts of the dossier. These arise from differences in regulatory practice and procedures, differences in practices of medicine and pharmacy, and differences in access to diagnostic and therapeutic procedures. What are the major differences in content and how can companies cope with them to make filings in the major developed world markets?
Module 1 Differences
Although the ICH CTD refers to Module 1 as comprising “Regional Administrative Information” (such as the application form, labeling, text of prescriber, and patient information), in practice there are other differences and some of key ones are summarized in table 2. It is usual for companies to prepare a common Core Data Sheet for information to health practitioners that could then be used to prepare draft Prescribing Information and Patient Information for the United States, the Summary of Product Characteristics (SmPC), and Patient Information Leaflet for the European Union.
|
Nation/region |
Key differences |
|
European Union |
1.4 Information about the Experts (who sign the Module 2 Summaries) |
|
1.5 Specific Requirements for Different Kinds of Application (summaries to support |
|
|
1.6 Environmental Risk Assessment |
|
|
1.7 Information relating to Orphan Market Exclusivity |
|
|
1.8 Information relating to Pharmacovigilance |
|
|
1.9 Information relating to Clinical Trials |
|
|
United States |
1.3.5 Patent and exclusivity information |
|
1.9 Pediatric administrative information |
|
|
1.16 Risk management plans |
|
|
Japan |
Patent status |
|
Background of origin, discovery, and development |
|
|
List of related products |
|
|
Data for review of designation as poisons, deleterious substances, etc. |
|
|
Draft of basic protocol for postmarketing surveillance |
|
|
Canada |
1.2.4 Patent information |
|
1.4 Health Canada Summaries |
|
|
1.5 Environmental Assessment Statement |
|
|
Australia |
1.4.1 Information about the Experts (who sign the Module 2 Summaries) |
|
1.5 Specific requirements about different types of application (literature based, |
|
|
1.6 Drug and Plasma Master Files and Ph Eur Certificates of Suitability |
|
|
GMP clearance letters |
|
|
Summary Biopharmaceutic Studies |
|
|
Pediatric Development Program |
|
|
Environmental Risk for non-GMOs containing medicines |
|
|
Antibiotic resistance data |
CPID: This is a condensed summary of current and specific chemistry and manufacturing information attested by the manufacturer and sponsor. There is no requirement for Expert signatures for the Module 2 Summaries in registration filings in the United States, Japan, and Canada.
Module 2 Differences
Although the formal ICH requirements for the Module 2 Summaries are identical in all ICH
and other countries, there are likely to be national or regional differences particularly in elation to the contents of the Module 3: Quality and Module 5: Clinical Studies modular files, and these will be reflected in these high-level summaries. For example, where a pharmacopoeial drug is the subject of a DMF, there will be just a reference to the DMF in Module 2 of a U.S. or Japanese dossier, whereas in the European Union there will be a summary of the Open Part of the European Union Active Substance Master File (DMF).
Module 3 Differences
Some of the key national or regional differences in content of Module 3 in relation to the drug substance and drug product are summarized in Table 3 and Table 4.
In addition, there are the 3.2.R Regional Differences. Examples quoted in the ICH CTD
are as follows:
- Executed Batch Records for Drug Substance and Drug Product (the United States only): Copies of records with equipment specified, manufacturing conditions, packaging records, and batch reconciliation information (theoretical yield, actual yield, and packaged yield).
- Method validation package for drug substance and drug product (the United States only)
- Comparability protocols (the United States only)
- Process validation scheme (the European Union only), including a process validation protocol where validation studies on the manufacturing process for the drug product are not complete
- Medical device used in combination with the drug product (the European Union only)
In addition, highly abbreviated documentation (a “lite” document) on the manufacture of the drug substance in 3.2.S.2.2 Description of the Manufacturing Process and on 3.2.P.3.3 Description of the Manufacturing Process and Process Controls for the drug product may need to be supplied in countries where regulatory agencies do not always respect the confidentiality of data.
There may also be differences in marketing needs for different countries. Thus, blister or foil packs are usually the packaging material of choice for tablets or capsules in the European Union whereas in the United States high-density polyethylene (HDPE) bottles are much more commonly used. In such a case, 3.2.P.7 Container Closure will include different information and also the primary stability data in 3.2.P.8 Stability for the European Union will be in blister or foil packs and in the United States it will be in HDPE bottles.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table 3: Summary of Some Key National or Regional Differences in Module 3.2.S
|
Nation/region |
Key differences |
|
European Union |
3.2.S Drug substance data may be submitted as an EU 2-part DMF (Open Part to be reproduced in 3.2.S) or as a reference to a Ph. Eur Certificate of Suitability (for Ph |
|
3.2.S.7 Stability: Storage requirements to be stated in accord with CHMP guideline |
|
|
United States |
3.2.S Reference may be made in the dossier to DMF information supplied directly by the drug substance manufacturer to FDA |
|
3.2.S.7 Stability: Storage requirements to be stated in accord with FDA labeling |
|
|
Japan |
3.2.S Reference may be made in the dossier to DMF information supplied directly by |
|
Canada |
3.2.S Reference may be made in the dossier to DMF information supplied directly by |
|
Australia |
3.2.S Drug substance data may be submitted as a 2-part DMF (Open Part to be |
Table 4: Summary of Some Key National or Regional Differences in Module 3.2.P
|
Nation/region |
Key differences |
|
European Union |
3.2.P.1 Description and Composition: Colors to be on the European Union permitted list. Excipients to be designated as conforming to Ph Eur or a European national pharmacopoeia where there is a monograph. |
|
3.2.P.4 Excipients: To conform to Ph Eur/European national pharmacopoeia if described in a monograph. |
|
|
3.2.P.5 Control of Drug Product: Assay limits to be ± 5% unless justified. A different manufacturing and shelf-life specification may be required. |
|
|
3.2.P.7 Container Closure System: Name of manufacturer(s) not required unless product is critical (e.g., parenteral). |
|
|
3.2.P.8 Stability: Storage requirement to be in accord with CHMP guideline. |
|
|
United States |
3.2.P Reference may be made in the dossier to DMF information supplied directly to FDA by excipient and container/closure manufacturers. |
|
3.2.P.1 Description and Composition: Colors to be on FDA permitted list. Excipients to be designated as conforming to USP/NF where there is a monograph. |
|
|
3.2.P.4 Excipients: To conform to USP/NF if described in a monograph. |
|
|
3.2.P.5 Control of Drug Product: Assay limits allowed to be up to ± 10%. A single regulatory (shelf-life) specification is allowed. |
|
|
3.2.P.7 Container Closure System: Name of manufacturer(s) required. |
|
|
3.2.P.8 Stability: Storage requirement to be in accord with FDA requirements for wording. |
|
|
Japan |
3.2.P.1 Description and Composition: Colors to be on Japanese permitted list. |
|
3.2.P.4 Excipients: To conform to monographs of the Japanese Pharmacopoeia or Japanese Pharmaceutical Excipients. |
|
|
Canada |
3.2.P.7 Container/Closure: Reference may be made to a DMF from the supplier. |
|
3.2.P.8 Stability: Storage conditions to refer to Health Canada requirements (e.g., storage at controlled room temperature). |
|
|
Australia |
3.2.P.1 Description and Composition: Colors to be on Australian permitted list for colors in oral products. |
|
3.2.P.8 Stability: Storage conditions to refer to TGA list of acceptable storage conditions (e.g., Store below 30◦C). |
Module 4 Differences
There are usually no major differences in this module.
Module 5 Differences
Some of the key differences are summarized in Table 5.
Table 5: Summary of Some Key National or Regional Differences in Module 5: Clinical Studies
|
Nation/region |
Key differences |
|
Europe |
Where required BE studies for generic products need to use a European batch of |
|
Clinical trials should normally comply with CHMP Efficacy guidances where these exist. |
|
|
Clinical trials of new drug products versus European authorized “gold standard treatment” are important as well as placebo studies |
|
|
United States |
Clinical trials should normally comply with FDA regulatory guidances where these |
|
FDA Integrated Summaries of Safety and Efficacy (ISS/ISE) to be included in 5.3.5.3 |
|
|
Japan |
“Bridging” pharmacokinetic and clinical studies may be needed to allow foreign data |
|
Other Non-ICH countries |
“Bridging pharmacokinetic and clinical studies may be needed to allow foreign data |
Managing the Differences
If companies wish to file simultaneously in a number of the major developed world markets, the chemical, pharmaceutical, nonclinical, and clinical development program needs to be designed to meet all of the individual market regulatory needs. For example, additional “bridging” pharmacokinetic or clinical trials may be needed to support a foreign registration file in Japan. Most of the documentation in Modules 2 to 5 for a major new drug registration file can be identical, but where there are national differences in requirements (e.g., differences in 3.2.P.5.1 Drug Product Specification in terms of assay limits for the European Union and U.S. markets), it is usually more efficient to prepare the two versions of the document at the same time. The Module 2.3 Quality Overall Summary and the Module 2.5 Clinical Overview could be prepared as identical “core documents” but they should then be reviewed by in-country staff and customized as necessary to meet any different technical or regulatory requirements of the different agencies.
ADOPTION OF THE CTD FORMAT OUTSIDE THE ICH REGION
The CTD format is being adopted with local modifications as needed by other national regulatory agencies and regional groupings of agencies. The Association of Southeast Asian Nations (ASEAN) comprises Brunei Darussalam, Cambodia, Indonesia, Laos, Malaysia, Myanmar, Philippines, Singapore, Thailand, and Vietnam. These countries have a combined population of over 550 million. They have published the ASEAN CTD (10). The ICH format is allowed for NCE and Biological products, but compliance is needed with ASEAN technical requirements [e.g., the ASEAN guideline on Stability Study of Drug Product (10)] and the ASEAN CTD (ACTD) administrative requirements. The ACTD will be implemented across the whole region by January 1, 2009.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
ASEAN COMMON TECHNICAL DOCUMENT [ACTD][19-22]
ASEAN Introduction
The Association of South-East Asians Nations (ASEAN) is a regional organization consisting of ten member countries, namely, Brunei Darussalam, Cambodia, Indonesia, Laos, Myanmar, Malaysia, Philippines, Singapore, Thailand and Vietnam.
ASEAN was established in 8 August 1967 by the governments of five countries - Indonesia, Malaysia, Philippines, Singapore and Thailand. In 1984, Brunei Darussalam joined its neighbors in the association. As a group, these early participants are dubbed the ASEAN6. Vietnam has become member since 1995, Laos and Myanmar since 1997 and Cambodia since 1999. These four new members are usually referred to as the CLMV group.
In 1992, the governments of ASEAN member countries agreed to create the ASEAN Free Trade Area (AFTA) to set common tariff scheme. The agreement on Common Effective Preferential Tariff Scheme (CEPT) has been effective since 2003. Pharmaceutical trade among the members now enjoy import duties of 0-5 % under CEPT, provided that the products has no less than 40% local content.
A Pharmaceutical Product Working (PPWG) was established to work on the details in the development of harmonization guidelines for technical procedures and requirements partially applicable to the ASEAN pharmaceuticals have been identified for harmonization - quality, efficacy, safety and administrative data.
Key documents resulted from work of PPWG include :
- ASEAN Common Technical Requirements (ACTR) for pharmaceutical product registration (for human use).
- ASEAN Common Technical Dossier (ACTD) for pharmaceutical product registration (for human use).
- ASEAN Guidelines on the following areas : analytical validation, bioavailability and bioequivalence studies, process validation, stability.
While ASEAN was working on its harmonized pharmaceutical registration, another international collaboration for harmonization of pharmaceutical registration was taking place in parallel called the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). It was established in 1990 and works for development for technical guidelines for registration of pharmaceutical products to achieve greater harmonization.
ASEAN Pharmaceutical Product [24]
At the first PPWG meeting the terms of reference were agreed and it was decided that the topics selected for harmonization would be divided into safety, quality and efficacy to reflect the three criteria which would be the basis for approving medicinal products. One of the PPWGs key topic is the idea of an ‘ASEAN pharmaceutical product’. This means that same regulatory requirements apply for the registration of a medicinal product among the ASEAN member countries. The PPWG developed the ASEAN Glossary of terms, the ACTD, the ACTR and their guidelines.
The ACTD gives information on the format and structure of the dossier that shall be commonly used for applications in the ASEAN region. The ACTD should serve as a locator for documentation that has been compiled for a marketing authorization application. It does not give any recommendations on the actual content of the dossier. The ACTD is similar to the European Notice to Applicants Volume 2B Presentation and Format of the dossier (EU-CTD).
The ACTR is as set of written material intended to guide applicants to prepare an application in a way that is consistent with the expectations of all ASEAN Drug Regulatory Authorities. It is a guide for preparation of the ACTD.
For Pharmaceuticals, efforts to develop harmonization schemes of pharmaceutical regulations in ASEAN to facilitate trade in pharmaceuticals continued and for this ATCD, covering administrative data, quality, safety and efficacy and an ASEAN Common Technical Requirements (ATCRs), covering quality, safety and efficacy had developed. The ACTD is the part of marketing authorization application dossier that is common to all ASEAN member countries while the ATCR is the set of written materials, intended to guide applicant(s) to prepare application dossiers in a way that is consistent with the expectations of all ASEAN Drug Regulatory Authorities. Series of guidelines for the implementation of the ATCR are being finalized. The ASEAN Standards and Quality Bulletin is regularly published with a view to ensure dissemination of information and promote transparency on standards, technical regulations and conformity assessment procedures in ASEAN member countries.
Regulatory Agencies in South East Asia
Table 6 : List of countries and their Regulatory Authority (ACTD) [19-20]
|
Country |
Regulatory Agencies |
|
Indonesia |
National Agency of Drug & Food Control |
|
Malaysia |
Drug Control Authority, NCE Unit |
|
Philippines |
Department of Health |
|
Thailand |
Thai FDA Drug Control Division |
|
Singapore |
Health Sciences Authority (HSA) |
|
Brunei |
Ministry of Health |
|
Vietnam |
Drug Administration of Vietnam |
|
Myanmar |
Food and Drug Administration |
|
Cambodia |
Department of Drug, Food and Cosmetics |
|
Laos PDR |
Ministry of Health, Food And Drug Department (FDD) |
ORGANIZATION OF ASEAN CTD (ACTD) FORMAT [23]
ACTD is a guideline for the preparation of a well-structured CTD applications that would be submitted to ASEAN regulatory authorities for the registration of pharmaceuticals for human use. This guideline describes a CTD format that would significantly reduce the time and resources needed to compile applications for registration and in the future, would ease the electronic document submissions.
Four parts are :
- Part I : Table of Contents, Administrative Data and Product Information
- Part II : Quality Document
- Part III : Nonclinical Document
- Part IV : Clinical Document
Presenting above information in the organizational structure which is known as ACTD Triangle.
The organizational structure is detailed with the help of technical data of ASEAN common technical document (ACTD) which is mentioned in all part of respective dossier.

upon request
Fig. 2 Organizational Structure of ACTD
This format is explained with selected drug Alogliptin for Singapore.
Overview of ICH-CTD and ACTD
In this thesis an attempt has been made to compare the drug regulatory approval procedure and requirements for the registration of pharmaceuticals for human use in ICH countries and ASEAN. The main points of divergence are in the content and format of the registration dossier.
The ACTD consists of Parts I to IV which have subsections A to F whereas ICH-CTD has 5 Modules with subsections that are numbered. The administrative data of Part I is part of ACTD whereas Module 1 of ICH-CTD is purely country specific. The summaries of the quality (Part II), nonclinical (Part III) and clinical (Part IV) are located at the beginning of each part of the ACTD. The ICH-CTD dedicates these summaries in a separate Module 2. As the ACTD does not have such summary part, it consists of only 4 Parts. The major differences between the ICH-CTD and ACTD are listed below in Table.
Table 7 : DOCUMENTS LOCATION IN FORMAT OF ICH-CTD & ASEAN CTD [13]
|
ICH CTD |
ASEAN CTD |
Description |
Remarks |
|
Module 1 -Regional and Administrative Information |
Part I |
Contains documents that are specific to each region. This module is not part of CTD. Basically consists of administrative documents like Application form, legal documents (GMP, Licenses etc.), labeling etc. |
Required for generics and New Drug. |
|
Module 2 - Overall Summary |
Incorporated in Parts II, III and IV |
This module summarizes the Module 3, 4 and 5. It includes Quality Overall summary, Non Clinical Overview and Summary and Clinical Overview and Summary. The summary provides reviewer the abstract of documents provided in the whole application. |
Required for generics and New Drug. For generics summary on Quality part only required. |
|
Module 3 - Quality |
Part II |
The documents related to Chemistry, manufacturing and Control of both Drug Substance and Drug Product is included in this module. |
Required for generics and New Drug. |
|
Module 4 - Safety |
Part III |
Non Clinical Study Reports – Data on pharmacologic, pharmacokinetic, and toxicological evaluation of the pharmaceutical product is provided. |
Not required for generics. |
|
Module 5 -Efficacy |
Part IV |
Clinical Study Reports - A critical assessment of the clinical data and related reports is provided in this module. |
Not required for generics except Bioequivalence study. |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Drug Profile [24-35]
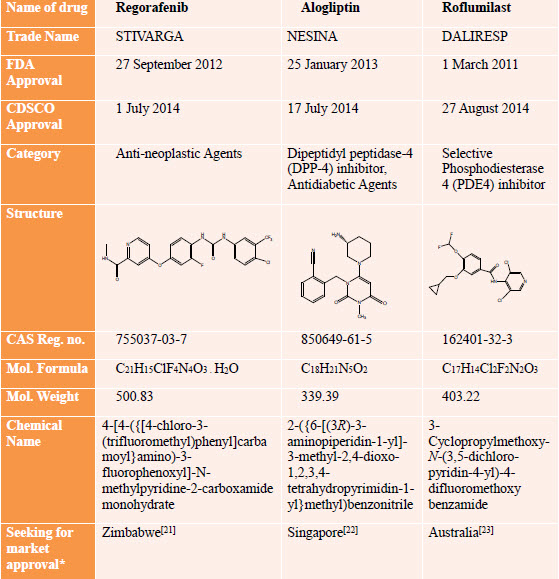
*As of May 2015
Conclusion
From the study it could be understood that getting a market authorization for registration of a drug in any territory requires a particular format, and that each country follows a specific guideline in addition to its own regulations which are laid down by the respective drug regulatory authority. There are basically two formats available in most of the countries of world one is , ICH-CTD and the other is ACTD. ICH-CTD is followed by ICH countries where as ACTD is usually adopted by ASEAN countries. The ICH-CTD has five modules, in which module 1 is country specific presenting administrative information of the country but the other four modules which are framed for drugs. They include descriptive scientific details information and are common to all the countries which follow the format. Module - 2,3,4,5 have summary of quality, nonclinical and clinical information, respectively. The ACTD has parts instead of module. They are four in number. These include summary of quality, nonclinical and clinical in part II, III, IV, respectively.
References
1. Dictionary reference. https://dictionary.reference.com/browse/dossier?s=ts.(accessed August 2014).
2. World Health Organization Homepage www.apps.who.int/prequal/info.../GenericGuideline_PDS_CTD-Format.pdf. (accessed August 2014).
3. World Health Organization Homepage www.apps.who.int/prequal/info_general/documents/ WHO_DMP_RGS_98_5_R.pdf. (accessed August 2014)
4. Pharma Tutor Homepage
https://www.pharmatutor.org/articles/registration-dossier-pharmaceuticals. (accessed August 2014).
5. Central Drug Standard Control Organisation Homepage www.cdsco.nic.in/writereaddata/CTD%20Guidance%20Final.pdf. (accessed August 2014).
6. International Journal of Pharmaceutical Sciences Review and Research Homepage https://www.globalresearchonline.net/journalcontents/volume9issue2/Article-030.pdf. (accessed August 2014).
7. World Health Organization Homepage
https://www.who.int/en/ (accessed August 2014).
8. Pan American Health Organization (PAHO) Homepage
https://www.paho.org/hq/ (accessed August 2014).
9. World Trade Organization (WTO) Homepage
https://www.wto.org/ (accessed August 2014).
10. ICH Homepage https://www.ich.org/home.html (accessed August 2014).
11. Spark Pharma Regulatory Consultant Homepage https://www.sparkpharmareg.com/pharmaceutical_regulatory_affairs.html (accessed September 2014).
12. Perfect Pharmaceutical Consultant Pvt. Ltd. Homepage
https://www.perfectdossier.com/ctd-dossiers.html (accessed September 2014).
13. Regulatory Affairs Professionals Society (RAPS) Home Page https://www.raps.org/Regulatory-Focus/News/Databases/2015/04/06/21908/The-Essential-List-of-Regulatory-Authorities-in-Asia/ (accessed September 2014).
14. ICH Homepage
https://www.ich.org/products/ctd.html (accessed September 2014).
15. ICH Homepage https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4_R3_ Organization/M4_R3__organization.pdf. (accessed September 2014)
16. ICH Homepage https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4_R1_ Quality /M4Q__R1_.pdf. (Accessed September 2014).
17. ICH Homepage https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/ M4__R2__Safety/M4S_R2_.pdf. (accessed September 2014).
18. ICH Homepage https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4__R1__Efficacy/M4E__R1_.pdf. (accessed September 2014).
19. Association of Southeast Asian Nations (ASEAN) Homepage
https://www.asean.org/ (accessed September 2014).
20. Health Science Authority (HSA) Singapore Homepage https://www.hsa.gov.sg/content/hsa/en.html (accessed September 2014).
21. Institute of Developing Economies Japan External Trade Organization Homepage https://www.ide.go.jp/English/Publish/Download/Report/2008/pdf/2008_0111_ch3.pdf. (accessed September 2014).
22. Asean Development Bank Homepage https://www.adb.org/sites/default/files/publication/156295/adbi-wp440.pdf. (accessed October 2014).
23. Ministry of Health Brunei Darussalam Homepage www.moh.gov.bn/pharmacyservices/download/ASEAN%20Common%20Technical%20Document%20(ACTD).pdf. (accessed October 2014)
24. Bayer: Science for better life - Canada. www.bayer.ca/files/STIVARGA-PM-ENG-2OCT2013-159750.pdf. (accessed October 2014).
25. U.S. Food and Drug Administration Homepage https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm321378.htm. (accessed October 2014).
26. Central Drug Standard Control Organisation Homepage https://www.cdsco.nic.in/forms/default.aspx. (accessed October 2014).
27. Central Drug Standard Control Organisation Homepage https://www.cdsco.nic.in/forms/list.aspx?lid=1820&Id=11. (accessed October 2014).
28. U.S. Food and Drug Administration Homepage https://www.fda.gov/safety/medwatch/safetyinformation/ucm355781.htm. (accessed 0ctober 2014).
29. U.S. Food and Drug Administration Homepage https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203085lbl.pdf. (accessed 0ctober 2014).
30. Drug bank Homepage https://www.drugbank.ca/drugs/DB08896. (accessed November 2014).
31. Drug bank Homepage https://www.drugbank.ca/drugs/DB06203. (accessed November 2014).
32. Drug bank Homepage https://www.drugbank.ca/drugs/DB01656. (accessed November 2014).
33. Medicine Control Authority of Zimbabwe. https://www.mcaz.co.zw/. (accessed November 2014)
34. Center for Pharmaceutical Administration Health Sciences Authority - Singapore https://www.hsa.gov.sg/content/hsa/en.html. (accessed November 2014)
35. Therapeutic Goods Administration - Australia. https://www.tga.gov.au/. (accessed November 2014).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






